
AI-Powered Compliance Platform for Life Sciences
April 24, 2026

Expanded access (also called compassionate use) is a regulatory pathway that allows patients with serious or life-threatening conditions to receive investigational drugs outside of clinical trials when no comparable approved therapy exists and the patient cannot enroll in an ongoing trial. In the US, the FDA administers three expanded access pathways — individual patient INDs (including emergency use approved by phone in under 24 hours), intermediate-size patient protocols, and treatment INDs for broader populations. In the EU, expanded access operates through national competent authorities under Article 83 of Regulation 726/2004, with significant variation by member state.
Expanded access programs are not clinical trials — they exist solely for treatment, not research — but they carry real regulatory obligations including IND maintenance, adverse event reporting, and IRB/ethics oversight. For sponsors, the calculus is strategic as well as ethical: FDA analysis shows that drugs with expanded access experience have a higher approval rate (84%) than those without (76%), and EA data is increasingly used in regulatory submissions and health technology assessments. This guide covers when sponsors should offer expanded access, how the US and EU frameworks differ, and the operational requirements for running a compliant program.
Expanded Access (also known as Compassionate Use) is a U.S. FDA and EU regulatory framework that enables patients to obtain investigational drugs when:
Key Difference from Clinical Trials: Clinical trials generate safety and efficacy data under controlled conditions, while expanded access is strictly for treatment purposes, not research.
Right-to-Try legislation (2018) provides a separate pathway, allowing access without FDA oversight or IRB review, but requires that the drug has completed Phase I trials and that the manufacturer agrees.
Sponsors may offer EA when:
Many sponsors manage expanded access alongside broader patient support operations. Zelthy's patient services platform handles both, with enrollment, eligibility screening, adherence monitoring, and engagement workflows on a single platform.
EA pathways accommodate various development stages, though specific requirements vary by program type.
Sponsors must provide safety data showing:
The FDA evaluates whether “risks are not unreasonable given the disease severity.”
Three main EA pathways exist under FDA:
European programs operate through national competent authorities under Article 83 of Regulation 726/2004. The EMA provides harmonized recommendations, but implementation varies by member state.
For example, countries like:
These safety monitoring and audit trail requirements are part of a broader compliance landscape. Zelthy's compliance and quality platform automates adverse event lifecycle management, inspection readiness, and continuous GxP documentation.
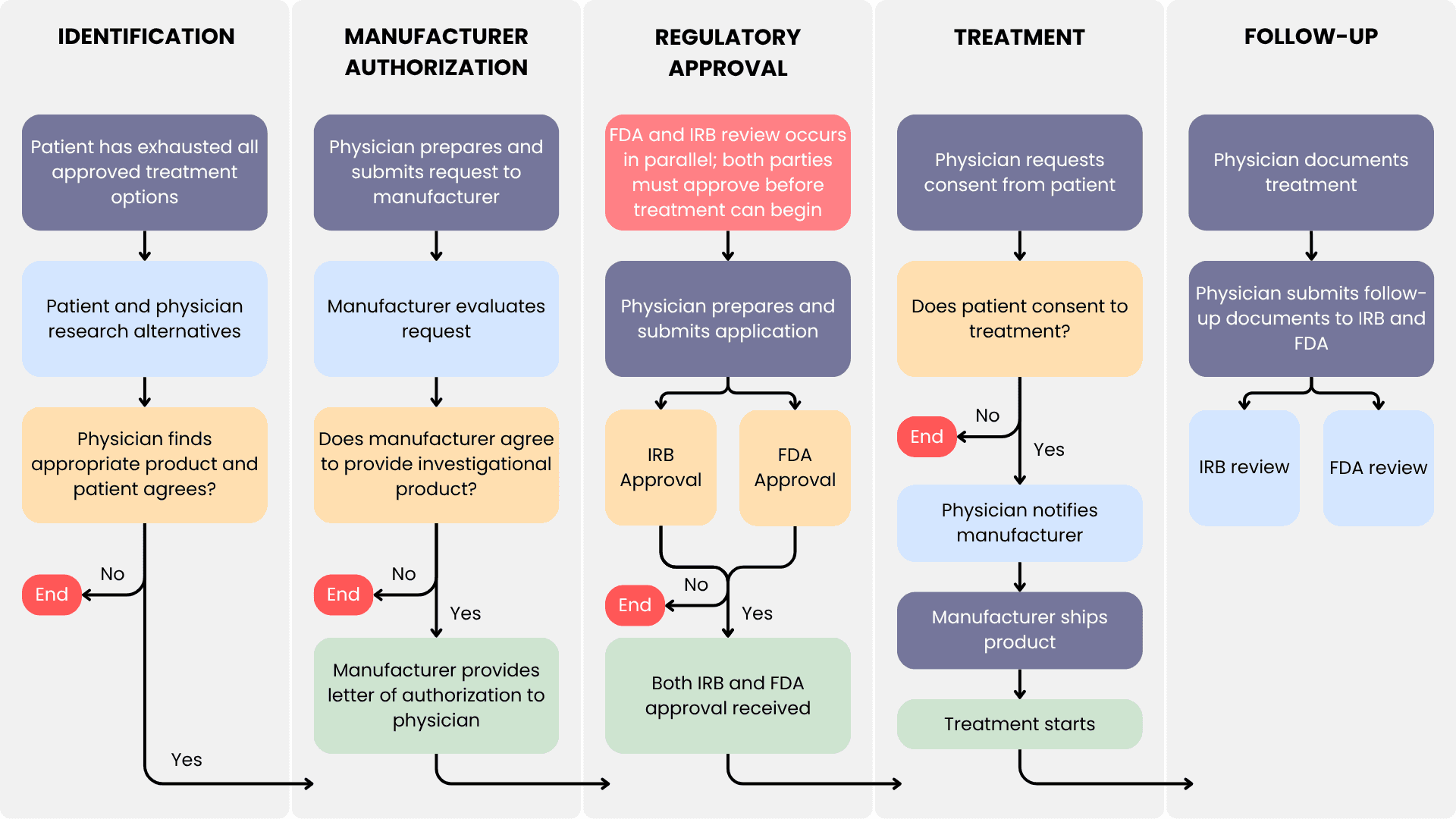
Expanded Access Request Process (Single Patient)
The single-patient expanded access process begins when a treating physician identifies a patient with a serious or life-threatening condition who cannot participate in available clinical trials. For emergency use, the FDA can authorize access by phone in under 24 hours, with IRB notified within 5 days. For non-emergency requests, both IND submission and IRB approval are required, with typical FDA review taking approximately 4 days. The sponsor must provide current safety data and maintain the IND with protocol amendments, annual reports, and expedited safety reporting for serious adverse events within 15 days.

US expanded access operates under FDA oversight through three pathways: individual patient INDs (including emergency use with phone authorization in under 24 hours), intermediate-size patient protocols, and treatment INDs for broad populations. All require IND submission and IRB approval (except emergency use, where IRB is notified post-treatment within 5 days).
EU compassionate use operates through national competent authorities under Article 83 of Regulation 726/2004, with implementation varying by member state — France uses Temporary Authorization for Use (ATU), the UK offers the Early Access to Medicines Scheme (EAMS) with real-world data collection, and countries like Italy and Spain require ethics review while others delegate to physicians.
Both regions require safety monitoring and adverse event reporting, but US reporting obligations are more standardized (15-day expedited reporting for serious events) while EU requirements vary significantly by country.
These US–EU regulatory differences mirror the broader divergence in how each region designs patient support. Our US vs EU PSP comparison maps these structural differences across the full patient services landscape.
Managing Expanded Access requires precise coordination across regulatory submissions, patient eligibility tracking, safety reporting, and supply management. Many sponsors—especially emerging biopharma—struggle to balance these demands alongside pivotal clinical trials.
Zelthy’s digital health platform simplifies this process by:
By integrating operational, regulatory, and patient-facing functions into a single platform, Zelthy enables sponsors to expand patient access without straining resources—while maintaining compliance, transparency, and data integrity.
Expanded access management is one module within Zelthy's broader regulatory operations platform — which also covers submission pipeline management, dossier assembly, regulatory intelligence, and labelling management across jurisdictions.
A leading Australian pharmaceutical company partnered with Zelthy to digitize its Compassionate Access Program (CAP), cutting approval times by 65%, reducing costs by 40%, achieving 99% compliance with automated audit trails, improving real-time visibility for stakeholders, and strengthening supply chain forecasting—ultimately delivering critical therapies faster and more efficiently.
Read the full story to find out how they managed to cut approval times from 7–10 days to 2–3 days by digitizing their compassionate access program while maintaining full regulatory compliance.
Connect with us and talk to us about building your Expanded Access framework with Zelthy. For further inquiries, reach us at connect@zelthy.com or send us a DM on LinkedIn.
Expanded access, also called compassionate use, is a U.S. FDA and EU regulatory pathway allowing patients with serious or life-threatening diseases to access investigational drugs outside of clinical trials when no approved treatment exists. Unlike clinical trials, which generate safety and efficacy data under controlled conditions, expanded access is strictly a treatment pathway for patients who cannot enroll in a trial and have no other options.
Expanded access requires FDA authorization and, for non-emergency cases, IRB approval before a patient can receive an investigational drug. Right-to-Try legislation (2018) allows access without FDA oversight or IRB review, provided the drug has completed Phase I trials and the manufacturer agrees to supply it. Right-to-Try removes regulatory gatekeeping but also removes the FDA's patient safety oversight and adverse event monitoring requirements that apply to expanded access programs.
Sponsors may offer expanded access when a patient has a serious or immediately life-threatening disease, no comparable approved treatment exists, the patient cannot enroll in an available clinical trial, and the potential benefit justifies the risks. Individual patient access generally requires Phase I safety data. Intermediate-size and Treatment IND programs require progressively more robust safety and efficacy evidence before the FDA will authorize broad access.
For emergency individual patient requests, the FDA can grant approval by phone in under 24 hours. The sponsor submits the IND and IRB notification follows within five days of treatment. For non-emergency individual patient requests, typical FDA review time is approximately four days. This speed reflects the agency's recognition that patients with immediately life-threatening conditions cannot wait for standard regulatory timelines.
FDA analysis of 321 regulatory decisions found no instances where expanded access experience led to a negative approval outcome. In fact, drugs with expanded access programs have a higher approval rate (84%) than those without (76%), likely because real-world EA data supplements the clinical evidence package for health technology assessments and regulatory submissions. The primary concern for sponsors is operational, resource demands on regulatory, medical, and supply chain teams running EA alongside pivotal trials.
Since 2017, the FDA has approved multiple drugs based on real-world evidence, and expanded access data increasingly contributes to this evidence base. EA data, capturing treatment outcomes, adverse events, and dosing patterns outside trial conditions, supports HTA submissions, post-market safety reporting, and comparative effectiveness arguments in payer negotiations. Sponsors who implement structured data collection in EA programs, rather than treating them as purely operational, generate regulatory and commercial value from patient access commitments.
In the EU, compassionate use is governed by national competent authorities under Article 83 of Regulation 726/2004, meaning program structure and approval timelines vary by country. France uses Temporary Authorizations for Use (ATU); the UK operates the Early Access to Medicines Scheme (EAMS). The EMA provides harmonized recommendations but does not centrally authorize programs. By contrast, the US FDA runs a unified expanded access framework with standardized IND submissions and defined review timelines applicable across all states.
![AI Output Compliance Monitoring in Pharma: The Watch Layer [2026]](/_next/image?url=https%3A%2F%2Fhumble-friendship-ab99f71d93.media.strapiapp.com%2FAI_Compliance_in_Pharma_a76c884d28.png&w=3840&q=75)
